Enfermedades raras
Síndrome de Goldenhar
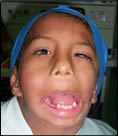
El síndrome de Goldenhar, es un síndrome muy raro polimalformativo congénito (que se evidencia en el nacimiento). La enfermedad se caracteriza por una amplia gama de síntomas y signos, que pueden variar mucho de un individuo a otro en función de la severidad del caso. En la mayoría de las ocasiones, la afectación es unilateral y asimétrica, de un lado más que del otro y suele afectar más al lado derecho aunque entre un 10% y un 33% de los afectados presentan alteraciones bilaterales.
Fue descrito por primera vez, en 1845, por Von Arlt, es clasificado como una entidad independiente en 1952 por Goldenhar y fue Gorlin en 1963, quien lo identificó como una displasia (desarrollo anómalo de tejidos u órganos) óculo-aurículo-vertebral.
Se desconoce la causa exacta de esta enfermedad que afecta al desarrollo del 1º y 2º arco branquial, pero se postula la existencia de un posible defecto, trauma o exposición intraútero a determinados factores ambientales. Apoyando esta teoría está el hecho de que este síndrome ocurra al parecer con mayor frecuencia en los hijos de Veteranos de la Guerra del Golfo. En pocos casos se asocia a un defecto genético.
Se trata de una enfermedad muy rara, cuya incidencia se estima en 1 de cada 25.000 nacidos vivos. Sin embargo, el trastorno genético que existe en los casos que aparecen de forma esporádica se presenta en 1 de cada 3.500 ó 5.000 recién nacidos vivos.
Afecta preferentemente a los varones con una relación V/M 3:2.
Las malformaciones suelen afectar a boca, oídos, ojos, pudiendo también localizarse en las vértebras:
- Se caracteriza por afectación facial con: hipoplasia (desarrollo incompleto o defectuoso) de las regiones malar, maxilar, y/o mandibular del lado afectado, también puede haber debilidad e hipoplasia de la musculatura facial en el mismo lado.
Puede asociarse con agenesia (desarrollo defectuoso, o falta de alguna parte de un órgano) unilateral de la parótida (glándula salivar de gran tamaño, situada en la cara por debajo y delante del oído. Su inflamación produce la parotiditis, también conocida como paperas) y la consiguiente disminución de la secreción de esta glándula. Se acompaña de micrognatia (mandíbula anormalmente pequeña), hipoplasia unilateral del paladar y/o músculos de la lengua, labio leporino (fisura del labio superior) y/o paladar hendido (cierre incompleto de la bóveda del paladar) presentes en el 7-15% de los pacientes, disfagia (dificultad para tragar) y apnea (ausencia o suspensión temporal de la respiración) del sueño.
- Las anomalías auriculares son: microtia (oreja muy pequeña, incluso reducida a diferentes apéndices) o anotia (ausencia congénita de una o ambas orejas), presencia de esbozos preauriculares fundamentalmente a nivel del trago, atresia (oclusión de una abertura natural) del canal auditivo y sordera; son bilaterales en alrededor de un tercio de los casos.
- Manifestaciones oculares: Tumores epibulbares, dermoides o lipodermoides , en el 35% de los pacientes, que son masas sólidas ovaladas, de color amarillento-rosáceo, mayores de 10 mm de diámetro, uni o bilaterales, que pueden dificultar la visión. Otras anomalías oculares acompañantes son: estrabismo (desviación de uno de los ojos de su dirección normal, por lo que los ejes visuales no pueden dirigirse en un mismo tiempo al mismo punto), microftalmía (ojos anormalmente pequeños) o anoftalmía (falta congénita de los ojos), blefarofimosis (hendidura palpebral corta) en el 10% de los casos, estrechamiento de la fisura palpebral, anomalías de la retina, colobomas (fisura congénita en alguna parte del ojo).
- Otras anomalías viscerales acompañantes son:
1.- Neurológicas: microcefalia (cabeza anormalmente pequeña) o hidrocefalia (acumulación de líquido en el encéfalo), encefalocele (salida del encéfalo a través de una abertura congénita o traumática del cráneo) occipital, malformación de Arnold Chiari, espina bífida (fisura congénita de los arcos vertebrales) y retraso mental en el 5-15% de los pacientes.
2.- Enfermedades cardiacas congénitas (presentes en el 5-58% de los casos): defectos del septo ventricular, ductus arterioso persistente, tetralogía de Fallot, coartación Aortica y transposición de los grandes vasos.
3.- Renales: ectopia (estado de un órgano o tejido, situado fuera de su lugar habitual) y/o fusión renal, agenesia renal, riñones displásicos multiquísticos, hidronefrosis (acumulo anormal de orina en los riñones), duplicación ureteral, reflujo vesico-ureteral, infecciones recurrentes del tracto urinario.
4.- Músculo esqueléticas:
a.- cabeza: hipoplasia de los huesos maxilar, temporal y malar, hipoplasia o ausencia de las ramas y cóndilos mandibulares, neumatización (presencia de gas en los tejidos) reducida de la región mastoidea;
b.-vertebrales: escoliosis (curvatura oblicua anormal de la columna dorsal), fusión de las vértebras cervicales en el 20-25% de los casos, platibasia (cráneo con base aplanada y ensanchada) y occipitalización del atlas en el 30% de los casos, anomalía de Klippel Feil, hipoplasia vertebral;
c.- otras anomalías esqueléticas: pie zambo (pie contrahecho), aplasia radial, anomalías del pulgar y anomalías costales.
El diagnóstico puede hacerse durante el embarazo mediante ecografía fetal y estudios genéticos. Posteriormente la ecografía del recién nacido así como los estudios radiológicos incluyendo escáner y la resonancia magnética nuclear permitirán concretar las malformaciones existentes, siendo las más frecuentes: agenesia y lipomas del cuerpo calloso, calcificación de la hoz del cerebro, hipoplasia del septum pellucidum y quistes dermoides intracraneales.
El diagnóstico diferencial debe hacerse con la microsomía hemifacial, enfermedad en la cual también existe una asimetría, pero limitada a la región cráneo facial.
Puede presentarse asociado a otros síndromes malformativos como el síndrome de CHARGE y el síndrome de Townes Brocks.
En cuanto al pronóstico, por lo general y dejando aparte los problemas derivados de las malformaciones, no suele acortar la vida ni la inteligencia, salvo que existan complicaciones.
Son imprescindibles los controles audiométricos (audiometría es la comprobación de la agudeza auditiva) precoces y periódicos y se debe hacer consejo genético.
El tratamiento suele ser de soporte y requiere una participación de un equipo multidisciplinario con pediatras, cirujanos plásticos y ortopedas, oftalmólogos, logopedas y demás especialistas implicados.
La corrección quirúrgica de las anomalías, debe indicarse precozmente, posponiendo la corrección de los defectos de la osteogénesis hasta alcanzar los dos años de edad. Las correcciones más frecuentes son canalizaciones para permitir la alimentación a través de la boca, reconstrucción del paladar o del labio, resección de los esbozos preauriculares, acortamiento o alargamiento de los huesos de la mandíbula, reconstrucciones malares, etc. La reconstrucción del oído externo puede requerir incluso tres o cuatro intervenciones.
Se discute la herencia en los casos de aparición familiar, aceptándose una heterogeneidad genética con patrones de herencia múltiple, autosómica dominante, recesiva o multifactorial, siendo el más frecuente el autosómico dominante.
Otras notas
Los científicos de las universidades de Liverpool y Glasgow han descubierto un posible nuevo método de mejorar la reparación de los nervios en el tratamiento de lesiones de la médula espinal.
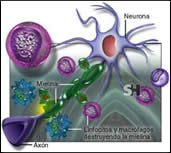
Es la enfermedad neurológica crónica más frecuente en adultos entre 20 y 40 años. Se estima que en todo el mundo más de 2.500.000 personas están afectadas por esta patología.
La familia es una estructura compleja que se mueve mediante reglas compartidas por todos sus miembros.